华中科技大学同济医学院刘恭平团队提出STAT3是治疗tau-N368诱导tau蛋白病...
华中科技大学同济医学院刘恭平团队提出STAT3是治疗tau-N368诱导tau蛋白病理的新分子标靶
既往研究证明信号转导和转录激活因子3(STAT3)在阿尔茨海默病中与调节突触可塑性、细胞凋亡和认知功能有关。来自中国华中科技大学同济医学院刘恭平团队在《中国神经再生研究(英文版)》(Neural Regeneration Research)上发表了题为“STAT3 ameliorates truncated tau-induced cognitive deficits”的研究中,通过将tau-N368以及STAT3腺病毒注射至小鼠海马CA3区,进一步揭示了在过表达天冬酰胺内肽酶对tau蛋白裂解产生tau-N368片段的小鼠模型中,STAT3对改善截短tau蛋白诱发认知障碍的分子机制。
阿尔茨海默病是一种随着时间推移逐渐破坏脑细胞的神经系统疾病,其病理特征包括过度磷酸化的tau蛋白形成的神经纤维缠结和β-淀粉样蛋白的细胞外积累形成的老年斑。Tau蛋白是一种主要的微管相关蛋白,可通过辅助微管组装和稳定,促进轴突运输并维持神经元功能。然而,在病理条件下,tau蛋白的过度磷酸化可促进和增强tau蛋白的聚集,从而在相关tau蛋白病中诱发神经退化。
天冬酰胺内肽酶,又称为δ-分泌酶,是一种溶酶体半胱氨酸蛋白酶,可在阿尔茨海默病大脑中被激活。这种酶存在年龄依赖性,可在N373和N585残基处裂解淀粉样前体蛋白,也可在N255和N368残基处裂解tau蛋白。其中Tau蛋白在N368残基处可被天冬酰胺内肽酶裂解导致该蛋白与微管结合的能力减弱,从而增强了其聚集能力以及神经毒性。截短的tau-N368片段比全长tau表现出更强的聚集能力和神经毒性,并可参与阿尔茨海默病的发生和发展。
STAT在阿尔茨海默病的病理过程中起着不可或缺的作用。一旦被激活,作为核内转录因子,STAT可在核内形成二聚体,并参与调节基因表达。众所周知,STAT与多种生物过程有关,如细胞增殖、分化、免疫调节、细胞生存和凋亡。刘恭平等既往研究表明,STAT1可在过表达人tau和人突变tau(htau和P301L-htau)的阿尔茨海默病小鼠模型中起到抑制N-甲基-D-天冬氨酸受体表达以及诱导突触和记忆障碍的作用[1, 2]。htau或P301L-htau的积累可通过诱导STAT1乙酰化和加强细胞质内STAT1和STAT3的相互作用而抑制STAT3转入细胞核。因此,过表达STAT3或非乙酰化的STAT1可通过上调N-甲基-D-天冬氨酸受体的表达,改善htau或P301L-htau诱导的突触可塑性损伤和认知障碍。
刘恭平等此次实验将tau-N368和STAT3质粒共转染进HEK293细胞,48h后,通过荧光素酶报告实验、电泳迁移率测定、Western blot分析和免疫荧光评估tau-N368的积累是否能够抑制STAT3转入细胞核,进而抑制STAT3的活性。然后利用脑立体定位注射法将tau-N368和STAT3腺病毒共同注射至2个月龄C57小鼠海马CA3区,应用水迷宫实验、巴恩斯迷宫实验、新事物识别实验和条件恐惧实验评估过表达STAT3是否改善tau-N368小鼠的认知功能障碍。最后通过电生理、蛋白免疫印迹、高尔基染色实验评估STAT3对tau-N368小鼠突触功能的改变,并通过蛋白免疫印迹、细胞凋亡检测试剂盒、免疫荧光等实验评估STAT3对tau-N368小鼠神经元的变化。结果表明,tau-N368的过表达可抑制STAT3转入细胞核的能力,使STAT3失去活性。而STAT3的过表达可改善tau-N368诱导的突触可塑性损伤,减少神经元的损失,从而改善其认知功能障碍。该研究结果也证实,当tau-N368在细胞内累积时,STAT3从细胞质到细胞核的转位受到抑制(图1)。在tau-N368小鼠中,可通过激活STAT3增加N-甲基-D-天冬氨酸受体蛋白水平,并降低Bax蛋白水平,进而成功逆转突触损伤和神经元损失,从而缓解tau-N368引起的认知障碍(图2-6)。总之,这一表明,STAT3可作为治疗tau-N368诱导的tau蛋白病理的新分子靶标。
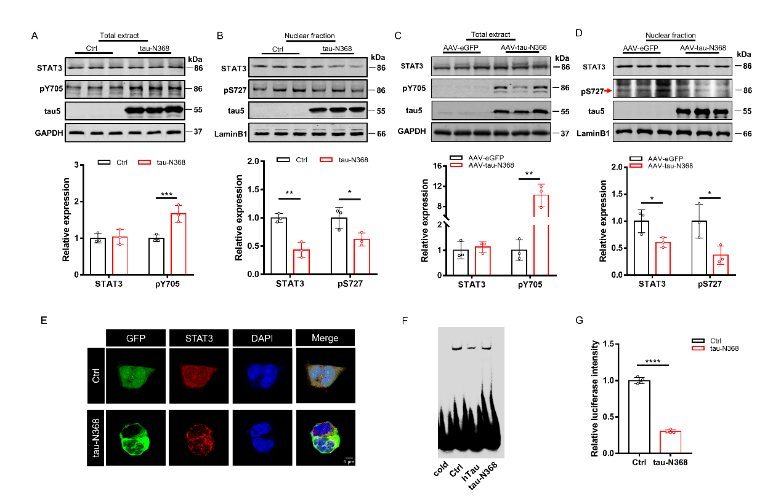
图1过表达tau-N368片段可通过抑制STAT3核转位导致STAT3失活(图源:Zhang et al., Neural Regen Res, 2024)
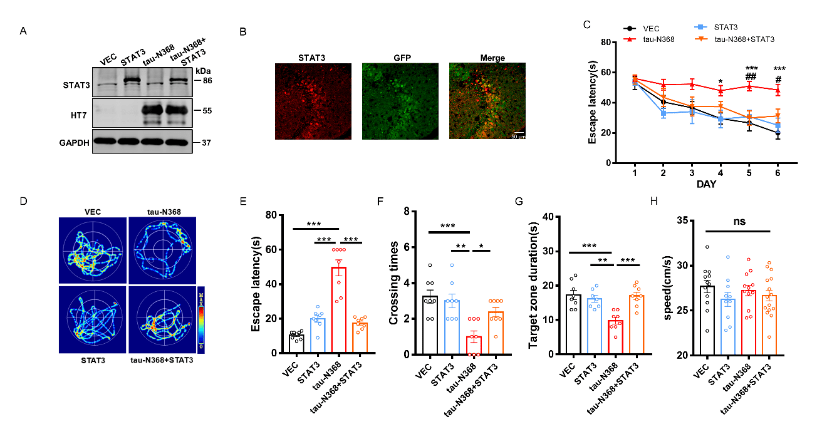
图2过表达STAT3可改善tau-N368诱导的认知障碍(图源:Zhang et al., Neural Regen Res, 2024)
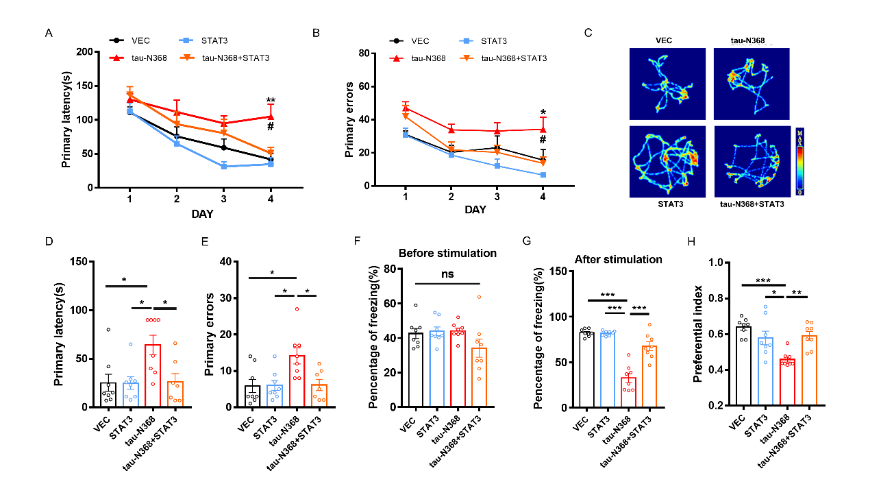
图3过表达STAT3可改善tau-N368诱导的认知障碍(图源:Zhang et al., Neural Regen Res, 2024)
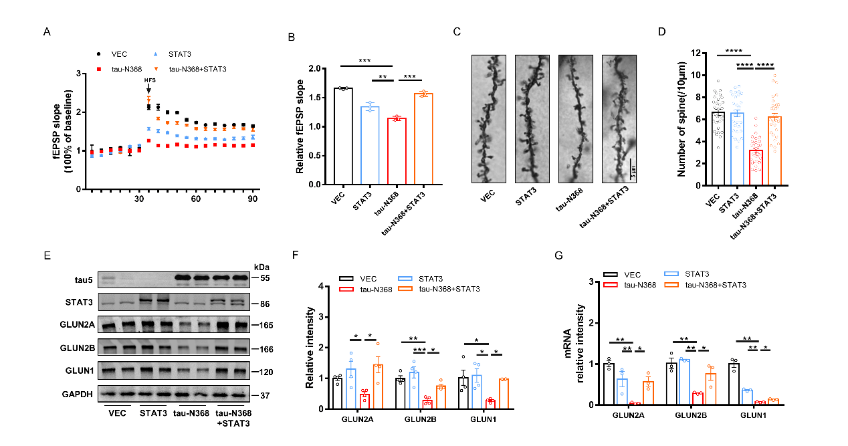
图4过表达STAT3可改善了tau-N368诱导的突触损伤(图源:Zhang et al., Neural Regen Res, 2024)
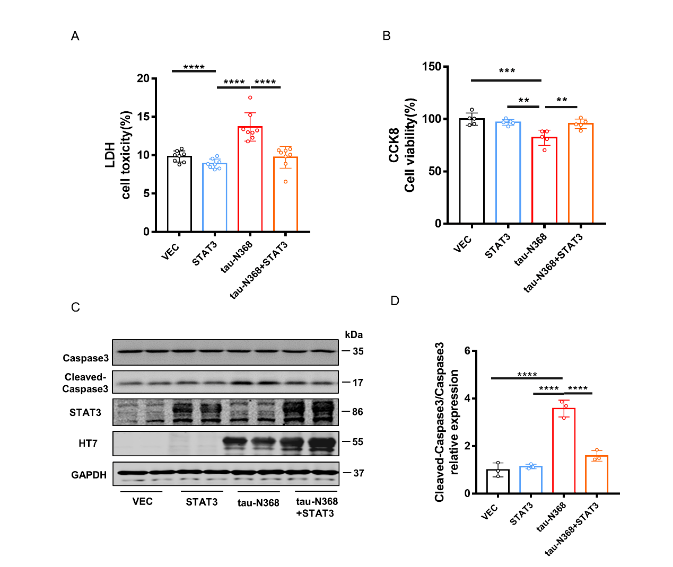
图5在体外过表达STAT3可改善tau-N368诱导的细胞毒性(图源:Zhang et al., Neural Regen Res, 2024)
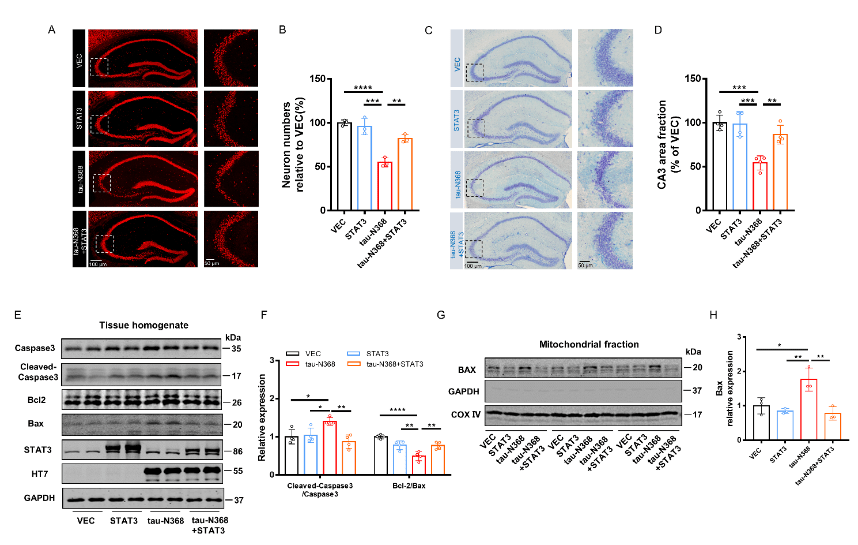
图6过表达STAT3可改善tau-N368诱导的神经元丢失(图源:Zhang et al., Neural Regen Res, 2024)
原文链接:https://doi.org/10.4103/1673-5374.382253
参考文献
[1] Wan HL, Hong XY, Zhao ZH, et al. STAT3 ameliorates cognitive deficits via regulation of NMDAR expression in an Alzheimer's disease animal model. Theranostics. 2021;11(11):5511-5524.
[2] Hong XY, Wan HL, Li T, et al. STAT3 ameliorates cognitive deficits by positively regulating the expression of NMDARs in a mouse model of FTDP-17. Signal Transduct Target Ther. 2020;5(1):295.

该研究由华中科技大学同济医学院博士研究生张兵歌、万华丽等共同完成。冯琼博士(右二)(华中科技大学同济医学院附属儿童医院)和洪小月博士(右一)(武汉大学附属中南医院)为共同通讯作者,华中科技大学为第一发表单位。
基金资助:国家自然科学基金青年科学基金项目(82101501,82201589)
文章摘要:天冬酰胺内肽酶切割tau蛋白产生的tau N368片段,可能驱动阿尔茨海默病患者大脑中与突触功能障碍和记忆退化相关的病理变化,但是截短tau诱导认知障碍的分子机制仍有待探索。有证据表明,信号转导和转录激活因子3(STAT3)与调节突触可塑性、细胞凋亡和认知功能有关。此项实验通过萤光素酶报告基因分析、电泳迁移率偏移分析、蛋白质印迹分析以及免疫荧光分析发现,在HEK293细胞中人tau-N368的积累可通过抑制STAT3核转运来抑制STAT3活性。而过表达STAT3可改善tau-N368诱导的突触可塑性损伤,减少神经元的损失,从而改善tau-N368小鼠的认知障碍。同时在tau-N368小鼠中,可通过激活STAT3增加N-甲基-D-天冬氨酸受体蛋白水平,降低Bax蛋白水平,进而逆转突触损伤和神经元损失,从而缓解tau-N368引起的认知障碍。综上,STAT3对截短tau相关神经病理变化起着重要作用,这提示了一种tau-N368影响突触和记忆障碍的新机制,并且STAT3可作为治疗tau-N368诱导的tau蛋白病理的新分子靶标。
文章关键词:阿尔茨海默病;神经退行性疾病;tau-N368;STAT3;突触;N-甲基-D-天冬氨酸受体;记忆;认知障碍;神经元缺失;细胞凋亡
文章来源:Zhang B, Wan H, Maierwufu M, Liu Q, Li T, He Y, Wang X, Liu G, Hong X, Feng Q (2024) STAT3 ameliorates truncated tau-induced cognitive deficits. Neural Regen Res 19(4):915-922.